Operate on clonotype tables¶
JoinSamples¶
Joins several clonotype tables together to form a joint clonotype abundance table. Joint clonotype holds information on all clonotypes that match under a certain comparison criteria (e.g. identical CDR3nt and V segment), their samples of origin and corresponding abundances. At least two samples should be specified for this routine. For two sample case also consider using OverlapPair routine.
Attention
This is the most memory-demanding routine, especially for a large number of samples.
Command line usage¶
$VDJTOOLS JoinSamples \
[options] [sample1.txt sample2.txt sample3.txt ... if -m is not specified] output_prefix
Parameters:
| Shorthand | Long name | Argument | Description |
|---|---|---|---|
-m |
--metadata |
path | Path to metadata file. See See Common parameters |
-i |
--intersect-type |
string | Sample intersection rule. Defaults to aa. See Common parameters |
-x |
--times-detected |
integer | Minimal number of samples in which a clonotype should be detected to get to the final output. Default = 2 |
-p |
--plot |
Turns on plotting. See Common parameters | |
-c |
--compress |
Compressed output for clonotype table. See Common parameters | |
-h |
--help |
Display help message |
Tabular output¶
Summary table suffixed join.[value of -i argument].summary.txt
is created with the following columns.
| Column | Description |
|---|---|
| <first sample id > | Indicator for the first sample, either 0 or 1 |
| <second sample id > | Indicator for the second sample |
| … | |
| clonotypes | Number of clonotypes detected in all samples that have 1 indicator in a given row. |
Joint clonotype abundance table file having join.[value of -i argument].table.txt
suffix that contains joint clonotypes detected in at least -x samples.
Table structure is described in the section below.
Joint clonotype abundance table structure¶
First columns have the same meaning as in VDJtools format clonotype abundance table, they are computed as follows:
Normalized frequency is computed as geometric mean of clonotype frequencies that comprise a given joint clonotype in intersected samples. If clonotype is missing, its frequency is set to
1e-9.Note
Joint clonotype is formed as a union of all clonotype variants in all samples that match under the specified
-irule.Normalized count is calculated by scaling normalized frequencies so that the joint clonotypes with smallest frequency has a count of
1.Clonotype signature (CDR3nt, CDR3aa, V, D and J) is taken from a representative clonotype.
Note
When several clonotype variants are present in samples that correspond to the same clonotype under
-irule (e.g. several Variable segment variants when-i ntis set), only the most abundant form is selected as a representative clonotype to final output.
| Column | Description |
|---|---|
| count | Normalized clonotype count |
| freq | Normalized clonotype frequency |
| cdr3nt | Representative CDR3 nucleotide sequence |
| cdr3aa | Representative CDR3 amino acid sequence |
| v | Representative Variable segment |
| d | Representative Diversity segment |
| j | Representative Joining segment |
| peak | Index of a time point at which given clonotype reaches its maximum frequency |
| occurrences | Number of samples the joint clonotype was detected in |
| <sample name> | Frequency of a joint clonotype at corresponding sample |
| … |
Graphical output¶
A Venn diagram can be found in a file having
join.[value of -i argument].venn.pdf suffix. Note
that if there are more than 5 samples, it will be
constructed for the first 5 samples. Plotting is performed
using VennDiagram
R package.
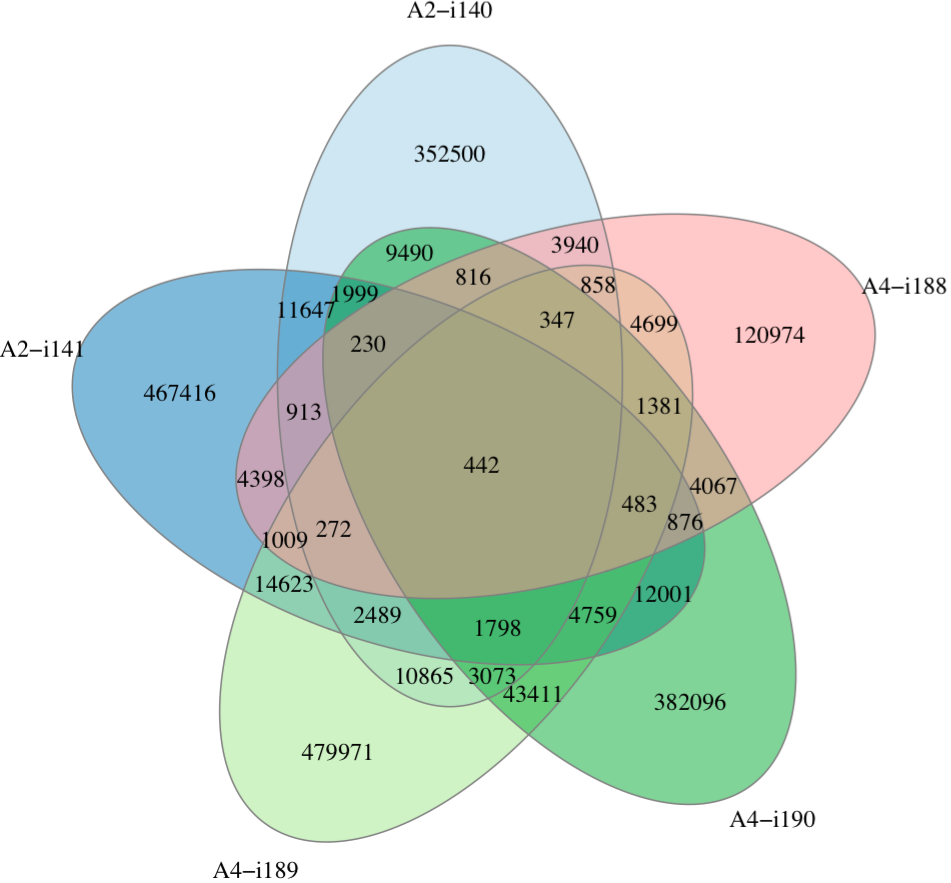
Overlap of clonotype sets. See Venn diagram wiki article for the description.
PoolSamples¶
Pools clonotypes from several samples together and merges clonotypes that that match under a certain comparison criteria (e.g. identical CDR3nt and V segment). Note that this routine can be used with a single sample to aggregate the sameple, e.g. by CDR3 amino acid sequence, in this case CDR3 nucleotide sequence, V and J segments will be taken from a representative clonotype variant with the highest frequency.
Command line usage¶
$VDJTOOLS PoolSamples \
[options] [sample1.txt sample2.txt sample3.txt ... if -m is not specified] output_prefix
Parameters:
| Shorthand | Long name | Argument | Description |
|---|---|---|---|
-m |
--metadata |
path | Path to metadata file. See Common parameters |
-i |
--intersect-type |
string | Sample intersection rule. Defaults to strict. See Common parameters |
-p |
--plot |
Turns on plotting. See Common parameters | |
-c |
--compress |
Compressed output for clonotype table. See Common parameters | |
-h |
--help |
Display help message |
Tabular output¶
Summary table suffixed pool.[value of -i argument].summary.txt
is created with the following columns.
| Column | Description |
|---|---|
| incidence.count | Indicator for the first sample, either 0 or 1 |
| read.count | Total number of reads associated with a given pooled clonotype |
| convergence | Total number of clonotype variants that match the pooled clonotype under -i rule. |
Pooled clonotype abundance table file having pool.[value of -i argument].summary.txt.
Table structure is described in the section below.
Pooled clonotype abundance table structure¶
First columns have the same meaning as in VDJtools format clonotype abundance table, they are computed as follows:
- Pooled count is computed as the total number of reads associated
with clonotype variants that match under the specified
-irule. - Frequency is computed as pooled count divided by total number of reads in all samples.
- Clonotype signature (CDR3nt, CDR3aa, V, D and J) is taken from a representative clonotype in the same way as described for Joint clonotype abundance table structure.
| Column | Description |
|---|---|
| count | Pooled clonotype count |
| freq | Pooled clonotype frequency |
| cdr3nt | Representative CDR3 nucleotide sequence |
| cdr3aa | Representative CDR3 amino acid sequence |
| v | Representative Variable segment |
| d | Representative Diversity segment |
| j | Representative Joining segment |
| incidence | Number of samples containing clonotype variants that comprise a given pooled clonotype |
| convergence | Total number of clonotype variants that match the pooled clonotype under -i rule |
Graphical output¶
planned